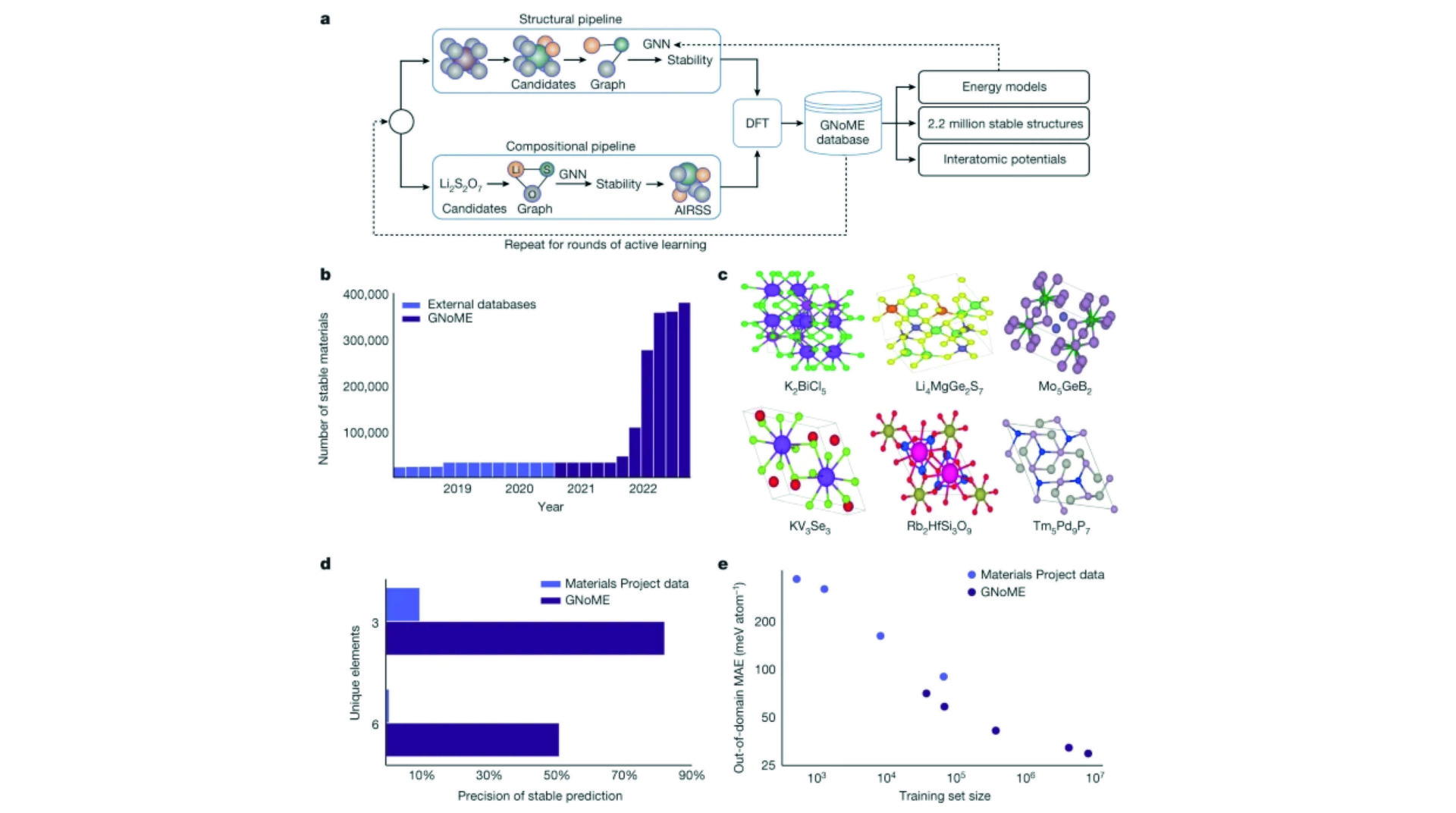
Scaling deep learning for materials discovery
Novel functional materials enable fundamental breakthroughs across technological applications from clean energy to information processing. From microchips to batteries and photovoltaics, discovery of inorganic crystals has been bottlenecked by expensive trial-and-error approaches. Concurrently, deep-learning models for language, vision and biology have showcased emergent predictive capabilities with increasing data and computation. Here we show that graph networks trained at scale can reach unprecedented levels of generalization, improving the efficiency of materials discovery by an order of magnitude. Building on 48,000 stable crystals identified in continuing studies, improved efficiency enables the discovery of 2.2 million structures below the current convex hull, many of which escaped previous human chemical intuition. Our work represents an order-of-magnitude expansion in stable materials known to humanity. Stable discoveries that are on the final convex hull will be made available to screen for technological applications, as we demonstrate for layered materials and solid-electrolyte candidates. Of the stable structures, 736 have already been independently experimentally realized. The scale and diversity of hundreds of millions of first-principles calculations also unlock modelling capabilities for downstream applications, leading in particular to highly accurate and robust learned interatomic potentials that can be used in condensed-phase molecular-dynamics simulations and high-fidelity zero-shot prediction of ionic conductivity.